Organická analýza - Principy plynové chromatografie

Foto: 2 Theta: Organická analýza
3 PLYNOVÁ CHROMATOGRAFIE
3.1 Principy plynové chromatografie
- 3.1.1 Parametry elučního profilu
- 3.1.2 Charakteristiky plynově chromatografického systému
- 3.1.3 Charakteristiky rozdělení
- 3.1.4 Optimalizace rozlišení
- 3.1.5 Identifikace analytů
- 3.1.6 Stanovení analytů
3.2 Schéma instrumentálního uspořádání GC
- 3.2.1 Mobilní fáze
- 3.2.2 Nástřik vzorku
- 3.2.3 Separační systém
3.3 Měřící systém
- 3.3.1 Signál v GC
- 3.3.2 Zpracování signálu
- 3.3.3 Detektory v plynové chromatografii
Kniha obsahuje přehled metod analýzy organických látek: Analytikům prohloubí jejich znalosti používaných metod a vedoucím pracovníkům poskytne podklady pro řešení úkolů jejich laboratoře. Je určena také pro studenty a vyučující univerzit a vědecké pracovníky.
💡 Kompletní obsah naleznete v odborné publikaci Organická analýza, kterou můžete zakoupit přímo u vydavatele 2 THETA, prostřednictvím LabRulez nebo v mnoha knihkupectvích.
Principy plynové chromatografie
Plynová chromatografie nese své označení podle skupenství mobilní fáze, kterou je plyn.
Využívá rozdělení koncentrace analytu mezi stacionární a mobilní fázi na základě adsorpce a rozpouštění, přičemž se předpokládá, že toto rozdělení je rovnovážné. Jako zdroje pohybu mobilní fáze využívá tlakový spád a stacionární fáze je uspořádána v koloně.
Analytická metoda plynové chromatografie využívá výše popsaný chromatografický děj sledovaný měřicím zařízením, jehož signál je funkcí množství analytu a citlivosti, ke kvalitativnímu a kvantitativnímu určení analytu. Analytické určení analytu se řídí pravidly stanovení nejistoty měření a tato pravidla zpětně určují stav, použití a vývoj metody plynové chromatografie.
Parametry elučního profilu
Eluční profil plynově chromatografického píku má obecně tvar exponenciálně modifikovaného normálního rozdělení a je obecně popsán analýzou momentů profilu.
Vrchol elučního profilu odpovídá maximálnímu množství analytu v integračním časovém úseku.
Šířka elučního profilu je definována druhým statistickým momentem, který odpovídá varianci (rozptylu) σ2 kolem těžiště profilu, tj. části plochy elučního profilu způsobenou variabilitou symetrických a exponenciálních dějů.
Experimentální šířka píku w neodpovídá pouze vlastnímu separačnímu ději, ale je tvořena řadou příspěvků k rozmytí koncentračního profilu od nástřiku vzorku až po detekci separovaných zón.
Takto způsobené rozmytí se projevuje pouze na klesající části elučního píku (za předpokladu, že sorpční isotherma není konkávní) a označuje se jako chvostování (ang. tailing).
Charakteristiky plynově chromatografického systému
Vlastnosti chromatografického systému nezávisejí na druhu analytu a jsou charakterizovány dvěma parametry. Jsou to schopnost opakovaného ustavení rovnováhy (účinnost systému) a schopnost ovlivnit velikost
rozdělovacího poměru (selektivita systému). Chromatografické podmínky, při kterých se nemění hodnota rozdělovacího poměru analytu, označujeme za jednodimensionální. Vícedimensionální chromatografie
potom odpovídá uspořádáním, při kterých se mění hodnota rozdělovacího poměru analytu (např. teplotní program, spojení kolon s různými stacionárními fázemi apod.).
Selektivita systému v plynové chromatografii je určena pouze vlastnostmi stacionární fáze (mobilní fáze je indiferentní plyn) a to jejími vlastnostmi absorpčními a zastoupením slabých interakcí (dispersní, polarizační,
přenos náboje).
Charakteristiky rozdělení
Smyslem analytické metody plynové chromatografie je rozdělení směsi analytů s nejmenší zbytkovou nejistotou. Pro posouzení stupně rozdělení v plynové chromatografii se používá několik kriterií, která lze
hierarchicky seřadit do následující řady:
rozlišení dvojice analytů, vyjádřené rozlišením Ri,j (viz kniha),
počet píků mezi dvěma sousedními n-alkany, vyjádřené separačním číslem SN100 (viz kniha),
rozsah retečních indexů (viz rov. 3.30) zahrnutých v jednom píku, vyjádřené informačním obsahem I(S) (viz kniha),
celková píková kapacita (d)-dimensionálního separačního systému nC(d) (viz kniha).
Tyto pojmy jsou hierarchicky uspořádány od dvojice analytů ke směsi analytů ve vzorku, od jednodimensionálního k vícedimensionálnímu systému a čím větší hodnotu dosahují, tím větší je stupeň rozdělení.
Optimalizace rozlišení
Identifikace analytů
Separační děj má dvě složky, kauzální a pravděpodobnostní. Kauzální část je tvořena chemismem chromatografického děje a transportními ději, kterým odpovídá retenční pořadí a účinnost separace.
Pravděpodobnostní část separačního procesu je spojena se zbytkovou nejistotou separace, tj. otázkou, zdali eluční pík odpovídá jen jedné látce.
Identifikace analytu i v analytické metodě plynové chromatografii, tzn. určení jeho nezaměnitelnosti s analytem j, vychází:
z určení jeho retence
a porovnání s retencí čisté látky,
výpočtem retenčních charakteristik a porovnáním s tabelovanými hodnotami,
a porovnání s teoretickou hodnotou použitím metod QSRR (quantitative-structure-retention relationships),
a z určení případných dalších nezaměnitelných molekulárních vlastností (viz detektory).
Jelikož absolutní retenční data jsou funkcí experimentálních podmínek (složení stacionární fáze, fázového poměru, teploty, tlakového spádu, průtoku mobilní fáze) používají se pro identifikaci z retenčních dat relativní
kritéria.
Stanovení analytů
Analytická metoda plynové chromatografie se používá pro velmi široký rozsah koncentrací analytů, počínaje určením hlavních složek směsi (>10%) až ke stopovým množstvím řádu zlomků ppb (tabulka 3.3). Hlavní oblast
aplikací je v koncentračním rozsahu analytů 0,1 až 10 ppm.
Schéma instrumentálního uspořádání GC
Základní schéma jednodimensionálního plynového chromatografu je tvořeno šesti samostatnými funkčními částmi, tak jak je ukázáno na obrázku 3.2 Samostatnost těchto funkčních částí spočívá nejen v jejich
zaměnitelnosti mezi jednotlivými výrobci (plynové chromatografy jsou často vyráběny v tzv. modulové technologii), ale i v nezávislosti fyzikálně chemických a inženýrských principů jejich konstrukce a funkce.
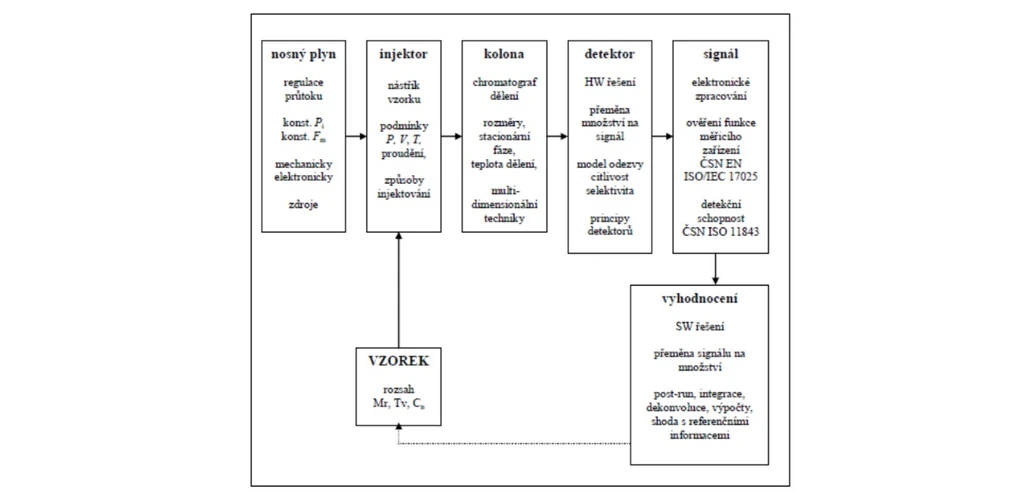 2 THETA: Obr. Schéma zapojení funkčních částí plynového chromatografu; Mr je relativní molekulová hmotnost, Tv je teplota
2 THETA: Obr. Schéma zapojení funkčních částí plynového chromatografu; Mr je relativní molekulová hmotnost, Tv je teplota
Mobilní fáze
Účinnost chromatografického systému je vedle míry interakčních vztahů analyt-stacionární fáze určena průměrnou lineární rychlostí mobilní fáze. V daném experimentálním uspořádání (konstantní průřez kolony A) je okamžitá lineární rychlost u mobilní fáze úměrná objemové rychlosti kolonou FC nastavitelné regulátory konstantního průtoku nosného plynu.
Jelikož plyny jsou stlačitelné, není okamžitá lineární rychlost plynu u konstantní po celé délce kolony, ale vzrůstá od počátku kolony k jejímu konci. Tento nárůst je významnější pro vyšší tlakový spád a obecně je určen
faktorem kompresibility j.
V analytickém uspořádání plynového chromatografu je regulován průtok nosného plynu na několika místech.
Aby nedošlo v detektoru k rozmytí píků v důsledku dlouhé doby setrvání analytu v detektoru, přidává se před detektorem dodatečné množství nosného plynu (Fmg), které zaručuje rychlé vymývání eluovaných analytů
z detektoru (Fdet). Za těchto podmínek dochází k dalšímu zředění okamžitého množství analytu, které může vést až k zániku vyhodnotitelného signálu (viz dále měřicí zařízení).
Průtok nosného plynu kapilární kolonou je popsán Poiseuilleovým zákonem. Základní tři uspořádání udržování konstantního průtoku v analytické metodě plynové chromatografie jsou: konstantní tlakový spád určený pouze kolonou, konstantní pneudynamický odpor sériového systému odporu (např. kapiláry) a kolony a konstantní hmotnostní průtok.
Regulátory konstantního vstupního tlaku jsou finančně nenáročné a v každém případě se doporučuje použít regulátor s kovovou membránou, neboť u regulátoru konstantního tlaku je jedna strana membrány otevřena proti okolní atmosféře (je nastavován přetlak proti okolí). Nečistoty, včetně vodní páry, přítomné v atmosféře se přes kovovou membránu nemohou dostat do nosného plynu. Levná řešení, používající k regulaci pouze externí regulovatelný odpor, např. jehlový ventil, namísto tlakového regulátoru, nejsou vhodná pro rutinní, přesnost vyžadující měření. Regulace konstantního vstupního tlaku je ovšem spojena s většími bezpečnostními riziky, neboť při nekontrolovaném přerušení vedení nosného plynu kdekoli za regulátorem dojde k úniku nosného plynu s vysokou průtokovou rychlostí a k vyprázdnění tlakové lahve do atmosféry
laboratoře.
Druhým typem regulátorů průtoku v plynové chromatografii jsou tzv. regulátory konstantního hmotnostního průtoku.
Mechanické regulátory konstantního hmotnostního průtoku jsou založeny na zpětné vazbě regulující tlak v děliči průtoku za regulátorem.
Mechanické regulátory konstantního hmotnostního průtoku vyžadují na vstupu regulovaný tlak P1 zpravidla o 100 kPa vyšší, než je výstupní tlak regulátoru Pi.
Elektronické regulátory konstantního hmotnostního průtoku jsou založeny na proporcionálním ventilu řízeném měřením teploty vlákna, která je funkcí tepelné kapacity nosného plynu.
Mobilní fáze v plynové chromatografii je plyn, nejčastěji vodík a helium, méně často dusík, argon, či oxid uhličitý. Zdrojem plynů jsou tlakové lahve. Tlakové lahve jsou plněny maximálně na 20 MPa a jsou označeny nezaměnitelnými barvami (vodík červeně, dusík zeleně, helium hnědě, vzduch bíle s černým pruhem, atd.) a nezaměnitelným lahvovým ventilem. Čistota plynů se označuje dvoučíslím X.Z, kde X je počet devítek a Z je následné významné číslo do stoprocentní čistoty (např. čistota označená 4.6 znamená čistotu 99,9960 %, tedy plyn obsahuje 40 ppm nečistot).
Pneumatická spojení v plynové chromatografii musí splňovat podmínku těsnosti a bezpečnosti při možném přetlakování. Skleněná a křemenná spojení, včetně kolon, se z tohoto důvodu nepoužívají do přetlaku vyššího než 400 kPa. Kovová spojení a kolony se nepožívají do přetlaku vyššího než 1500 kPa. Použití plastových spojení (včetně materiálu TEFLON®) a kolon není v plynové chromatografii vhodné, a to hlavně z důvodu absorpčních a permeabilních vlastností těchto materiálů vzhledem k plynům a parám z okolní atmosféry (permeabilní standardizační zažízení jsou konstruována z plastů).
Nástřik vzorku
Metoda plynové chromatografie vyžaduje, aby všechny látky vstupující do dělicího systému byly v plynné fázi. Tento požadavek určuje jak rozsah analyzovatelných látek, tak i možnost jejich kvantifikování. Obecně lze
v podmínkách plynové chromatografie analyzovat látky, až do teploty varu Tv ≤ 800 ºC, počtu uhlíkových atomů v molekule ≤ 100 a relativní molekulové hmotnosti Mr ≤ 1600. Nejčastější aplikační rozsah se však
pohybuje ve spodní polovině maximálních rozsahů. Za výše zmíněných podmínek je plynová chromatografie potenciální metodou pro analýzu velkého počtu organických látek (knihovny hmotnostních spekter
identifikovaných chemických látek mají více než 1.000.000 položek).
Plynová chromatografie využívá především slabých interakcí analytu se stacionární fází, z nichž obecně největší podíl zaujímají disperzní interakce, méně polární/dipolární a jen výjimečně se uplatňují interakce přenosu
náboje (acidobazické). Tomuto zastoupení interakčních sil odpovídá i menší vhodnost použití plynové chromatografie pro skupiny organických látek s donor-akceptorovými vazbami.
Zadání, které určilo koncentrační rozsah a účel analýzy, nepřímo určilo i způsob vzorkování. Analytická metoda plynové chromatografie se používá pro analýzu:
plynných vzorků
- bez úpravy,
- po jejich zakoncentrování
- v kapalinách a následném dávkování plynné fáze nad kapalinou (headspace),
- v kapalinách a následném dávkování kapalné fáze,
- na tuhých sorbentech a následné tepelné desorpci (např. SPE, viz kap. 2),
- vymražením a následné tepelné desorpci,
kapalných vzorků
- bez úpravy přímým nástřikem nebo nástřikem plynné fáze nad kapalinou (headspace),
- po jejich převedení na těkavé sloučeniny chemickou reakcí (derivatizace),
pevných vzorků
- po jejich rozpuštění ve vhodném rozpouštědle,
- po jejich derivatizaci,
- po jejich kontrolovaném tepelném rozkladu, pyrolýze.
Výše uvedený výčet ukazuje nejen aplikační šířku metody, ale naznačuje i velký počet různých postupů při přípravě analytického vzorku. Jelikož každý dílčí úkol analytického postupu je spojen s nenulovou zbytkovou
nejistotou, jsou postupy s menším počtem kroků výhodnější. Následující tabulka ukazuje četnost jednotlivých zdrojů chyb analytického procesu.
Tabulka Četnost chyb při analytické metodě plynové chromatografie
Zdroj chyby / Četnost [%]
úprava vzorku / 36,7
kontaminace / 29,8
operátor / 17,0
dávkování vzorku / 16,5
Zvláštní kapitolu tvoří vzorkovací metody s obohacovacím stupněm (zakoncentrováním analytů), a to jak pro vzorky z plynné fáze, tak i pro vzorky z kapalné fáze.
Způsob dávkování vzorku je vždy podmíněn typem vzorku a analytickým zadáním. Z hlediska frekvence analýz jsou metody dávkování:
on-line pomocí dávkovací smyčky: tento způsob vzorkování vylučuje hlavní zdroje chyb, kterými jsou úprava vzorku, kontaminace a lidský faktor včetně přesnosti dávkování. Na druhé straně tento způsob vzorkování dovoluje i optimalizaci nejen postupu vzorkování, ale i samotného separačního procesu, např. délky kolony,
off-line pomocí odběru vzorku a jeho následné analýze na různých místech a v jiném čase. Tento postup je zatížen většími chybami než on-line metoda, a to především v důsledku velkého počtu operací před vlastní
analýzou.
Teplota injektoru v klasickém uspořádání se volí tak, aby při dávkování došlo k okamžitému vypaření vzorku a na žádných místech injektoru (povrch septa, povrch vnitřní části a případné jeho náplně) nedošlo k následné kondenzaci. Volená teplota injektoru odpovídá střední části injektoru, ve které se nachází liner a je zpravidla o 50 ºC vyšší než teplota kolony. Vyšší teplota injektoru není vhodná, protože prostřednictvím kovových spojů mezi injektorem a kolonou injektor ovlivňuje teplotu kolony, a tak i přesnost výsledků. Teplotní gradient se šíří jak směrem ke koloně, tak i směrem k septu, jehož druhá strana je ve styku s okolním prostředím.
Separační systém
Účinnost a selektivita jsou charakteristiky separačního systému. Selektivitní faktor α byl definován jako poměr retence zvolených standardních látek (např. pro isomerně selektivní stacionární fáze jsou
zvoleny p-xylen a m-xylen). Proto je selektivita závislá na výběru standardních látek a popisuje výsledek interakce solut-stacionární fáze a nikoli vlastnosti samotné stacionární fáze.
V plynové chromatografii je známo několik set různých stacionárních fází. Jejich variabilita je nejen v chemickém složení fází, ale v jejich makromolekulárních vlastnostech, způsobu deaktivace povrchu kolon,
způsobu zakotvení na nosiči, způsobu vpravení do kolon, atd. V současné době došlo k omezení výběru a k výhradnímu používání profesionálně připravených kolon.
Standardními stacionárními fázemi pro rozpouštěcí mechanizmus retence (nejrozsáhlejší aplikační pole) byly pro analytickou metodu plynové chromatografie zvoleny:
SQUALAN® (alifatický uhlovodík) jako nepolární fáze (v současné době je nahrazován teplotně stabilnějším polymerem dimethylsiloxanu),
CARBOWAX® (polymer ethylenglykolu) jako polární fáze (v současné době je nahrazován teplotně stabilnějším polymerem kyanopropylsiloxanu).
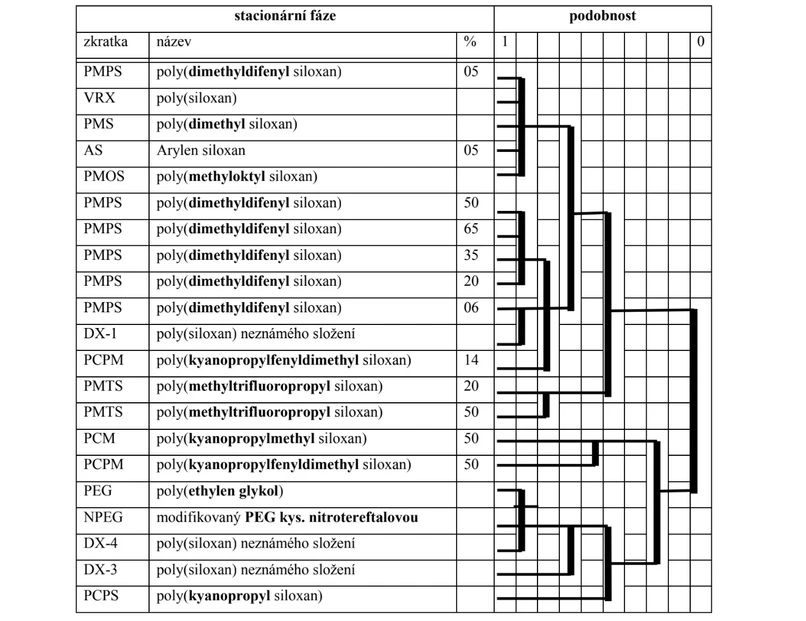 2THETA: Obr. Podobnost skupin stacionárních fází při 120 ̊C vyhodnocená hierarchickou klastrovou analýzou; % udává procenta polární funkční skupiny, podobnost 1 = identická, 0 = rozdílná
2THETA: Obr. Podobnost skupin stacionárních fází při 120 ̊C vyhodnocená hierarchickou klastrovou analýzou; % udává procenta polární funkční skupiny, podobnost 1 = identická, 0 = rozdílná
Zvláštní pozornost si v diskuzi stacionárních fází zaslouží zmínka o stacionárních fázích pro separaci opticky aktivních isomerů. Tyto fáze jsou zpravidla na bázi α-, β- a γ-cyklodextrinů, přičemž β-cyklodextrin je
používán nejčastěji.
Stacionární fáze je v koloně umístěna (používá se rovněž pojem zakotvena) buď na:
povrchu vhodného indiferentního nosiče, který naplňuje objem kolony zpravidla ze 70 %, tzv. náplňové kolony (packed column), nebo na
stěnách kolony ve formě tenkého filmu (open tubular column) o tloušťce vrstvy 0,1 až 1,0 μm, tzv. kapilární kolony nebo
vyplňuje objem kolony s výjimkou kapilárního volného prostoru, tzv. monolitické kolony.
Tloušťka vrstvy stacionární fáze na nosiči musí přitom být dostatečně velká (více než 0,2 μm pro nepolární a 0,5 μm pro polární stacionární fáze), aby byl vyloučen absorpčně-adsorpční retentenční mechanizmus
v mezifází nosič-stacionární fáze a na povrchu nosiče.
Měřicí systém
Obraz průběhu separace v koloně je zprostředkován měřicím zařízením, do něhož ústí výstup kolony. Měřicí zařízení se tedy neúčastní separace, ale využívá fyzikálních principů, které
převádějí množství analytu na elektricky měřitelnou veličinu označovanou jako signál (tzv. detektor) a
elektronicky zpracovávají signál.
Detektory v plynové chromatografii
Detektory jsou konstrukční části měřicích zařízení, ve kterých je fyzikálně chemická vlastnost analytu převáděna na měřitelný, zpravidla elektrický signál. Obecný přístup k posuzování detektorů je založen na diskuzi rovnice signálu (viz kniha); detektory jsou označovány podle využitelné vlastnosti ai, jejich citlivost a selektivita je popsána rovnicí (viz kniha). Analytická metoda plynové chromatografie se vyznačuje jak největší variabilitou detekčních možností, tak detektory s největší citlivostí a největší selektivitou mezi všemi separačními metodami. Tyto vlastnosti vyplývají z podstaty plynové chromatografie, kdy mobilní fází je
indiferentní plyn a z toho vyplývající amf ≈ 0 a Smatrice ≈ 0. Tabulka udává výběr detektorů často používaných v analytické metodě plynové chromatografie z hlediska interakčních energií částice a analytu
v detekčním prostoru. Tučně zapsané detektory jsou více popsány dále.
![2 THETA: Obr. Výčet detektorů používaných v analytické metodě plynové chromatografie; k udává hodnotu kolizní energie v [eV] pro ionizační detektory a emisní vlnovou délku v [nm] pro emisní
detektory](https://gcms.cz/labrulez-bucket-strapi-h3hsga3/2_THETA_Obr_Vycet_detektoru_pouzivanych_v_analyticke_metode_plynove_chromatografie_15bdea76c1/2-THETA-Obr-Vycet-detektoru-pouzivanych-v-analyticke-metode-plynove-chromatografie_l.webp) 2 THETA: Obr. Výčet detektorů používaných v analytické metodě plynové chromatografie; k udává hodnotu kolizní energie v [eV] pro ionizační detektory a emisní vlnovou délku v [nm] pro emisní
detektory
2 THETA: Obr. Výčet detektorů používaných v analytické metodě plynové chromatografie; k udává hodnotu kolizní energie v [eV] pro ionizační detektory a emisní vlnovou délku v [nm] pro emisní
detektory
Jak je ukázáno v tabulce, největší skupinu detektorů tvoří detektory ionizační. Jejich účinnost vyplývá ze srážkové teorie, která říká, že čím větší je energie a hmotnost částic, tím větší je účinný průřez kolize, s klesající molekulovým objemem klesá pravděpodobnost nepružné srážky (např. signál nulové linie bude klesat v řadě N₂ > H₂ > He), pravděpodobnost ionizace je největší v případě shody energie primární částice a ionizačního potenciálu analytu (např. energie fotonu značně vyšší než ionizační potenciál analytu nevede k jeho ionizaci), atd.
Detektor tepelné vodivosti (Thermal Conductivity Detector, TCD) měří změny tepelné vodivosti mobilní fáze způsobené přítomností eluované látky. Detektor má tedy dva paralelní kanálky, jeden referentní s konstantním průtokem nosného plynu a druhý měrný, který je připojen na výstup kolony. V kanálcích jsou umístěna žhavená tělíska ve formě drátků, drátků zalitých ve skle, termistorů nebo transistorů. Počet tělísek jsou dvě nebo čtyři, podle způsobu elektronického zapojení. Konstrukční detaily operátor nemůže měnit, jsou určeny výrobcem. Detekční objem se mění od 100 μL k několika jednotkám μL (tzv. micro TCD) a v případě leptaných řešení k několika nL.
Detektor elektronového záchytu (Electron Capture Detector ECD) byl uveden do analytické metody plynové chromatografie v roce 1960. Detektor je tvořen malou kovovou komůrkou, na jejichž vnitřních stěnách je umístěna fólie s β-zářičem. V komůrce jsou dvě sběrné elektrody a do komůrky ústí výstup kolony. Detekční objem ECD je kolem několika desítek μL (jeho zmenšování je omezeno brzdnou dráhou elektronů).
Konstrukční detaily detektoru, např. aktivita použitého zářiče, velikost polarizačního napětí na elektrodách, parametry frekvenčního uspořádání, atd. jsou určeny výrobcem a operátor je nemůže ovlivnit. Jelikož tento
detektor pracuje s radioaktivním materiálem, jsou pro něj uplatňována všechna kritéria práce s radioaktivními materiály. Servisní práce mohou být prováděny pouze vyškolenými firemními zaměstnanci.
Plamenově ionizační detektor (Flame Ionization Detector FID) je od svého zavedení v roce 1958 nejrozšířenějším detektorem v analytické metodě plynové chromatografie. Jeho podstata spočívá v měření
změny ionizačního proudu vodíko-vzduchového plamene způsobené přítomností eluovaného analytu. Plamenově ionizační detektor je tvořen hořákem, do kterého ústí výstup z kolony a přívod vodíku. Nad ústím
hořáku jsou umístěny sběrné elektrody iontů a elektronů vzniklých při hoření vodíku (velikost základního proudu bc je velmi malá) a analytu a dále přívod vzduchu pro difúzní plamen. Detekční objem plamenově
ionizačního detektoru je malý (5 až 10 μL) a je vhodný pro nejrůznější instrumentální uspořádání.
Plamenově fotometrický detektor (Flame Photometric Detector FPD) je založen na měření intenzity emise heteroatomů přítomných v molekule analytu. V současné době se FPD používá k selektivnímu měření fosfor obsahujících látek při vlnové délce 528 nebo 565 nm a měření síru obsahujících látek při vlnové délce 384 až 394 nm. Plamenový fotometrický detektor se v současné době nepoužívá pro měření látek obsahujících halogen. Detektor je tvořen hořákem, do kterého ústí výstup z kolony a přívod vodíku. Nad ústím hořáku je umístěn přívod vzduchu pro difúzní redukční plamen (teplota plamene je nízká a v plameni je přebytek vodíku) a světlovod. Detekční objem plamenově fotometrického detektoru je malý (10 až 30 μL), účinnost sběru světelného toku je předmětem různých konstrukcí.
Hmotnostně spektrometrický detektor (Mass Spectrometric detector, MS) je nejčastěji používaný v kombinaci s plynovou chromatografií. Řada jeho detekčních variant (Kap. 6) vede k vysoké selektivitě, a tak
ke snížení zbytkové nejistoty identifikace (viz rov. 3.28). MS je proto používán pro analýzy komplexních matric. Je nesporné, že současný technický vývoj činí MS detektor finančně dostupný a vede k postupné
náhradě ostatních selektivních detektorů v analytické metodě plynové chromatografie.
- [1] Ševčík Jiří, Měření a výsledky v analytické chemii, 2 THETA 2009, ISBN 978-80-86380-48-3
- [2] J. C. Giddings, Unified Separation Science, Wiley, New York 1991
- [3] J. Cazes, R. P. W. Scott, Chromatography Theory, M. Dekker, New York 2002
- [4] C. F. Poole, The Essence of Chromatography, Elsevier, New York 2003
- [5] V. Pacáková, K. Štulík, High Performance Liquid Chromatography, SPN, Praha 1990
- [6] K. Štulík, Analytické separační metody, Karolinum 2005
- [7] Analýza organických látek, 2 THETA, Český Těšín 2005, ISBN 80-86380-29-7



